

Discovery and Genetic Mapping of PM1, a Powdery Mildew Resistance Gene in Cannabis sativa L.
Paul D. Mihalyov* and Andrea R. Garfinkel
- 1Dewey Scientific, Pullman, WA, United States
- 2Oregon CBD, Independence, OR, United States
Powdery mildew is among the most common diseases of both hemp- and marijuana-type cultivated Cannabis sativa. Despite its prevalence, no documented studies have characterized sources of natural genetic resistance in this pathosystem. Here we provide evidence for the first resistance (R) gene in C. sativa, represented by a single dominant locus that confers complete resistance to an isolate of the powdery mildew pathogen Golovinomyces ambrosiae, found in the Pacific Northwest of the United States. Linkage mapping with nearly 10,000 single nucleotide polymorphism (SNP) markers revealed that this R gene (designated PM1) is located on the distal end of the long arm of one of the largest chromosomes in the C. sativa genome. According to reference whole genome sequences and Sanger sequencing, the marker was tentatively placed in a cluster of R genes of the nucleotide-binding site (NBS) and leucine-rich repeat (LRR) protein type. PM1's dominant behavior, qualitative penetrance, and a co-segregating qPCR marker to track its inheritance were confirmed in two separate genetic backgrounds totaling 185 recombinant F1 plants. The goal of this study is to provide a foundation for the discovery and characterization of additional sources of genetic resistance to pathogens that infect C. sativa.
Introduction
Emerging nutraceutical markets have driven increased agricultural interest in Cannabis sativa (2n = 2X = 20), commonly known as hemp or marijuana, due to its suite of secondary metabolites including the cannabinoids tetrahydrocannabinolic acid (THCA) and cannabidiolic acid (CBDA). Following the passage of the 2014 and 2018 Farm Bills that federally legalized the cultivation of hemp-type cannabis, there are now more than 100,000 acres grown across the United States (Olson et al., 2020). While traditionally bred plant varieties take multiple years to develop, the sudden onset and continued growth of cannabinoid demand has prompted the need for rapid structuring of hemp population genetics suitable for commercial cultivation. Among the many research focuses deserving attention, one such priority should be identifying genetic markers linked to naturally occurring disease resistance, thereby accelerating the development of varieties which are less dependent on the use of pesticides in disease management. Here we address the widespread need for collaborative germplasm improvement strategies by outlining the fastest route (single-cross) for mapping genetic resistance to powdery mildew, one of the most prevalent fungal diseases in cannabis.
Powdery mildew is a common term for several taxa of plant pathogenic fungi, including fungi from the genus Golovinomyces, which has been reported on both marijuana and industrial hemp-type C. sativa (Pépin et al., 2018; Szarka et al., 2019; Farinas and Peduto Hand, 2020; Maymon et al., 2020; Wiseman et al., 2021). Except for Wiseman et al. (2021) which uses the updated nomenclature, most of these reports identified the species G. spadiceus or G. cichoracearum sensu lato as the causal organisms, both of which have recently been included in the amended description of G. ambrosiae (Qiu et al., 2020). Golovinomyces ambrosiae is most widely reported on species of the Asteraceae family (Farr and Rossman, 2021); however, with the recent reassessment of the species, its host range may be much broader. Golovinomyces species represent a significant limitation to cannabis production, especially in greenhouse systems (Wiseman et al., 2021). Symptoms of powdery mildew include patchy white mycelial colonies which devhttps://www.frontiersin.org/articles/10.3389/fagro.2021.720215/full#B26elop on foliage and flowers, contributing to reduced plant vigor.
The economic impact of powdery mildew on cannabis production is largely unknown, or at least has not been quantified to date. While colony formation on the leaves or inflorescence does not necessarily result in total crop loss, infection can result in a reduction of end-use quality. Golovinomyces species have been found to be an important post-harvest contaminant of C. sativa. Microbiome analyses identified this genus on 15 of 19 samples of medicinal-grade marijuana purchased from dispensaries in northern California and was the third most abundant fungal genus found after the human pathogen-containing genus Purpureocillium and the cosmopolitan genus Penicillium (Thompson et al., 2017).
Fungicides may be used to manage powdery mildew, but are accompanied with uncertainties regarding compliance, efficacy, and consumer response. Regulatory agencies throughout the world have been slow to adopt new guidelines for the use of agrochemicals on cannabis, creating a backlog of research needed to safely apply pesticides. Furthermore, efficacy data for the use of pesticides against powdery mildew in cannabis is lacking (Scott and Punja, 2020). Alternative strategies to mitigate powdery mildew include strategic environmental control, the use of high-powered UV-C lamps (Scott and Punja, 2020), and application of plant-growth promoting rhizobacteria (Lyu et al., 2019), yet powdery mildew continues to represent one of the most prominent biological diseases in both field and greenhouse settings. Thus, the discovery and characterization of naturally occurring powdery mildew resistance genes are of vital importance to the growth and sustainability of the cannabis industry.
While there are anecdotal reports of genetic powdery mildew resistance in C. sativa, there have been no peer-reviewed studies on the heritability of this trait in hemp or marijuana to date (Stack et al., 2021). Among the derivatives of molecular plant-pathogen interaction models, crop domestication has favored single dominant R (resistance) genes that confer “vertical” or “qualitative” resistance to a generally narrower range of pathogen isolates (Kourelis and van der Hoorn, 2018). R genes conferring qualitative resistance have been implicated in the resistance of hop (Humulus lupulus), the most closely related species to C. sativa of economic importance, to powdery mildew (Henning et al., 2017; Padgitt-Cobb et al., 2019). Partial “horizontal” or “quantitative” resistance to powdery mildew, whereby the host initially becomes infected, but can limit further excessive colonization (Dyck and Kerber, 1985), has been documented in other crops. Several mechanisms have been implicated in partial resistance including loss-of-function mutations or deletions of families of genes that confer hyper-susceptibility, such as the mlo (mildew resistance locus O) gene family (Kusch and Panstruga, 2017).
Despite a wide range of diseases reported on C. sativa, we have not been able to identify any functionally documented R genes of any kind in this species. In this paper, we describe a naturally occurring powdery mildew R gene that was discovered by screening a Pacific Northwest germplasm collection. This gene, first observed in experimental line “PNW39,” confers complete resistance to a Washington State isolate of G. ambrosiae and was inherited in a dominant, single-gene fashion in two independent genetic backgrounds. Using a recently developed 40K SNP array, we successfully mapped the R gene in an F1 population and validated a qPCR-based marker for use in breeding and selection. SNP genotyping also allowed for the creation of a high-density linkage map to determine the relationship among markers physically located on the same chromosome. The results are reported herein.
Materials and Methods
Plant Material
Resistance to powdery mildew was first observed in photoperiod-sensitive experimental line “PNW39” after being grown in a greenhouse setting with high disease pressure. Two independent mapping populations were subsequently developed using parental material consisting of “PNW39,” susceptible and photoperiod-sensitive cultivar “Jumping Jack,” and a susceptible and photoperiod-insensitive recombinant inbred family “Dwy1337.” For the first population, feminized pollen from susceptible cultivar “Jumping Jack” was crossed to the female parent “PNW39.” The second population was developed by exposing female parent “PNW39” to an open pollination of multiple individuals from the “Dwy1337” family. At least 100 viable seeds were collected from each independent cross. Currently, the most prevalent accessions of C. sativa are highly heterozygous due to its natural preference for outcrossing and acute inbreeding depression (Onofri and Mandolino, 2017). Thus, the F1 families resulting from these hybridizations were genetically heterogeneous and allowed for linkage mapping of simple to moderately complex traits.
Disease Phenotyping
Eighty-nine F1 progeny from “PNW39” × “Jumping Jack” (Trial 1) and 96 F1 progeny from “PNW39” × “Dwy1337” (Trial 2) were grown within high-humidity (60–80%) enclosures under high pressure sodium light bulbs with daytime high temperatures of 25–28°C and nighttime low temperatures of 21–24°C. Infected plants serving as powdery mildew inoculants surrounded experimental plants for the duration of each trial. White colonies developed on leaves and stems of both mapping populations within 45 days of seedling germination. Plants were maintained through maturity (flowering) to confirm the integrity of resistance in all phenological stages. Plants were scored as “resistant” or “susceptible” since intermediate phenotypes were not apparent. Fifty-five clonally propagated individuals from the “PNW39” × “Jumping Jack” population were re-infected to provide replication and validation of initial scores. Thus, segregating phenotypic resistance was observed in three separate trials spanning two genetic backgrounds.
Pathogen Identification
To determine the species of powdery mildew causing disease, sequence data from the 5.8S and 28S rDNA and internal transcribed spacer regions 1 and 2 were generated. Mycelium was scraped lightly with a sterile pipette tip directly from the surface of infected leaf tissue and placed into a 0.2mL PCR tube containing: 10 μL Platinum Direct Universal Master Mix (Thermo Fisher), 8.4 μL dH20, and 0.8 μL of each of the primers ITS1-F (Gardes and Bruns, 1993) and TW14 (Mori et al., 2000; Takamatsu et al., 2013). PCR cycling conditions used were: initial denaturing at 94°C for 2 min followed by 35 cycles of 94°C for 15 s, 60°C for 15 s, and 68°C for 20 s. The PCR product was visualized on a 1.2% FlashGel™ DNA Cassette (Lonza), cleaned up using ExoSAP-IT Express (ThermoFisher Scientific), and sequenced (GENEWIZ) using the same primers used in PCR. Sequenced fragments were trimmed and aligned, and a consensus sequence was built using Geneious Prime (v. 2020.2.1). The consensus sequence was then compared to others in GenBank using the MegaBLAST tool (NCBI). No single spore isolate was developed for sequencing nor for resistance screenings; therefore, the results may potentially include a population of individuals of the same species.
Fungal structures were also observed using a light microscope (ECHO Revolve, ECHO, San Diego, CA). Measurements of 60 conidia and 5 conidiophores were made using the “Create Annotations” feature in the ECHO Revolve software. Photographs were also taken using the ECHO Revolve software.
Genotyping and Linkage Mapping
DNA was extracted from parents and progeny of the “PNW39” × “Jumping Jack” population using a Quick-DNA Plant/Seed Miniprep Kit (Zymo Research) following the manufacturer's instructions. High-density genome-wide markers were generated for all 91 samples (2 parents and 89 progeny) using an Illumina iSelect 40K SNP Array by Lighthouse Genomics (Vancouver, British Columbia, Canada). Sequencing was performed by Génome Québec (Montréal, Québec, Canada). DNA from the “PNW39” × “Dwy1337” population was extracted by the same method and used for marker validation experiments.
A total of 39,801 SNP markers were curated for the “PNW39” × “Jumping Jack” population, expressed as genotypes “AA,” “AB,” and “BB.” For quality filtering, the following types of markers were removed: (i) those with >10% missing data, (ii) those with monomorphic allele scores, and (iii) markers with no “AB” genotypes, as all polymorphic loci in this population structure should have heterozygous clusters.
The remaining markers were divided into three categories (B3.7, D1.10, D2.15) based on the notation described by Wu et al. (2002). Markers were characterized as type B3.7 (ab × ab) if both parental genotypes were heterozygous, then saved for linkage map construction if the progeny segregated in the expected ratio of 1:2:1 (aa:ab:bb) according to a chi-squared test. This resulted in a final total of 1,889 high-quality type B3.7 SNP markers. The first stage of genetic mapping in F1 populations includes generating separate linkage maps for each heterozygous parent; thus, the remaining markers were characterized as type D1.10 (ab × aa) or D2.15 (aa × ab). Markers were discarded if the progeny failed to reflect 1:1 segregation according to a chi-squared test. This resulted in a total of 3,779 type D1.10 markers and 4,220 type D2.15 markers.
Linkage maps were created in R (R Core Team, 2018) using the BatchMap (Schiffthaler et al., 2017) package and its dependency OneMap (Margarido et al., 2007), which are particularly useful for linkage map construction in outcrossing F1 populations, with the former using multi-threaded computing for memory intensive phasing and marker ordering algorithms. Our last filtering step utilized the function create.data.bins to minimize computing requirements by assigning only one representative bin to groups of redundant markers (those which exhibited zero recombination events between them). This resulted in a total of 2,051 unique high-quality SNP markers suitable for linkage map construction: 621 and 670 characterized as types D1.10 and D2.15, respectively, with 760 type B3.7 markers used to bridge the two linkage maps. Finally, a “dummy marker,” representing the binary powdery mildew responses, was included to elucidate its genetic linkage to our known SNPs. Linkage groups were established with a minimum Logarithm of the Odds (LOD) threshold of 10 and maximum recombination frequency of 0.35.
qPCR Assay Development and Marker Validation
A single locus, LH3804, was determined to predict the resistant phenotype with 100% accuracy in the “PNW39” × “Jumping Jack” population (see results). The probe sequence for this locus was provided by Lighthouse Genomics and used to search publicly available whole genome sequences from C. sativa to identify the physical location of the SNP. Publicly available genomes were queried using the MegaBLAST tool in the NCBI whole-genome shotgun contigs database; the genome for publicly available C. sativa cultivar “CBDRx” (GenBank assembly accession no. GCA_900626175.2) was downloaded and searched using the same tool implemented in Geneious Prime. Primers Mildewey_1F (5′ TGAGGATGATCCAATGCCAACA 3′) and Mildewey_1R (5′ ACGACAATGTTCATGAGACAACA 3′) were then designed to amplify an ~650 bp region surrounding the probe sequence. PCRs were carried out for the parents “PNW39” and “Jumping Jack” and selected progeny from both segregating populations in 20 μL reactions containing: 10 μL 2x PlatinumTM II Hot-Start PCR Master Mix (ThermoFisher), 0.8 μL of 5 μM each primer (Integrated DNA Technologies), molecular grade water, and 2 μL template DNA (variable concentrations). Cycling conditions used were initial denaturing at 94°C for 2 min followed by 35 cycles of 98°C for 5 s and 60°C for 15 s. PCR reactions were visualized as described above; all products were of the expected size. PCR cleanup, sequencing, and editing of sequence data were also as described above.
Sanger sequencing confirmed the presence of an A/G mutation in accordance with the SNP array at locus LH3804. Flanking region sequences were used to design a TaqMan ® SNP Genotyping Assay (ThermoFisher), which employed two primers (5′ TCATCCATCTATCTTGTGTTATTTCATTGCT 3′ and 5′ ACACTATCCTCTGATTCTGCATTCA 3′) and two probes (5′ /VIC/TGCATTTTCTGTGTTGGCAT/MGB-NFQ/ 3′ and 5′ /FAM/TGCATTTTCTGTATTGGCAT/MGB-NFQ 3′). This SNP assay was used to test progeny from both segregating populations in 10 μL qPCR reactions containing: 5 μL TaqPathTM ProAmpTM Master Mix (Applied Biosystems), 0.5 μL of the SNP Genotyping Assay (20x), 1μL of template DNA, and water. Samples were run in a QuantStudioTM 5 (Applied Biosystems) using the “Fast” and “Genotyping” protocol with the following cycling conditions: a pre-read at 60°C for 30 seconds; 95°C for 5 min; 40 cycles of 95°C for 5 s followed by 60°C for 30 s; and a post-read at 60°C for 30 s. Genotyping calls were made automatically by the QuantStudioTM 5 software and manually checked for accuracy.
Phenotyping of C. sativa Populations Suggests That a Single Dominant Gene Is Associated With Disease Resistance
Dense, powdery, white mycelial growth developed on susceptible individuals whereas resistant plants remained free of any visible signs of infection (Figure 2). Fungal colonies were not detected on “PNW39,” acting as a resistant control during each trial. Phenotyping the replicated subset of “PNW39” × “Jumping Jack” validated our results from the first trial (Supplementary Table 1).

FIGURE 2. Experiments with heavy disease pressure imposed through plant maturity revealed (A) susceptible phenotypes with dense mycelial growth (pictured: individual #45 from the population “PNW39” × “Jumping Jack”) and (B) resistant phenotypes absent of any powdery mildew colonies (pictured: individual #49 from the population “PNW39” × “Jumping Jack”).
A chi-squared test was conducted to evaluate progeny segregation against the null hypothesis of a single dominant Mendelian gene inherited from a heterozygous resistant parent. Both populations fit this assumption (Table 1), providing evidence that not only is there a single candidate locus, but its efficacy and heritability holds up in multiple genetic backgrounds. Full phenotype results are provided in Supplementary Tables 1, 2.
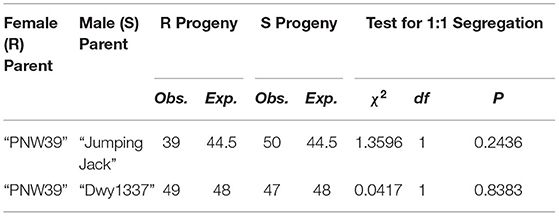
TABLE 1. Chi-squared tests for goodness of fit among each mapping population. To satisfy the null hypothesis, a 1:1 segregation is expected of resistant (R) to susceptible (S) individuals.
PM1 Co-localizes With SNP Marker LH3804 on Chromosome 2
A total of 2,051 SNP markers were anchored to 28 linkage groups (14 for each parent), some with as few as 8 markers. Since the C. sativa genome only has 10 pairs of chromosomes, the smaller linkage groups are likely artifacts of chromosomal regions with unconfirmed linkage to larger fragments. Here we focused on the 141-marker linkage group containing our phenotypic marker for PM1, which we have identified as chromosome 2 according to the “CBDRx” genome assembly (accession no. LR213632.1) (Supplementary Table 3).
Binary disease scores co-segregated with one type D1.10 SNP marker, locus LH3804, in 100% of progeny. The location of this marker bin (130.1/145.3 cM, or 95.39/96.15 Mb) places locus LH3804 in a distal position on the long arm of chromosome 2 of “CBDRx” (accession no. LR213632.1, formerly chromosome 6, accession no. UZAU01000623.1) (Figure 3 and Supplementary Table 3). The physical locations of the probe sequences for the additional markers on the linkage map largely corroborate their placement on “CBDRx” chromosome 2, except for proximal regions with low marker density (Supplementary Table 3).
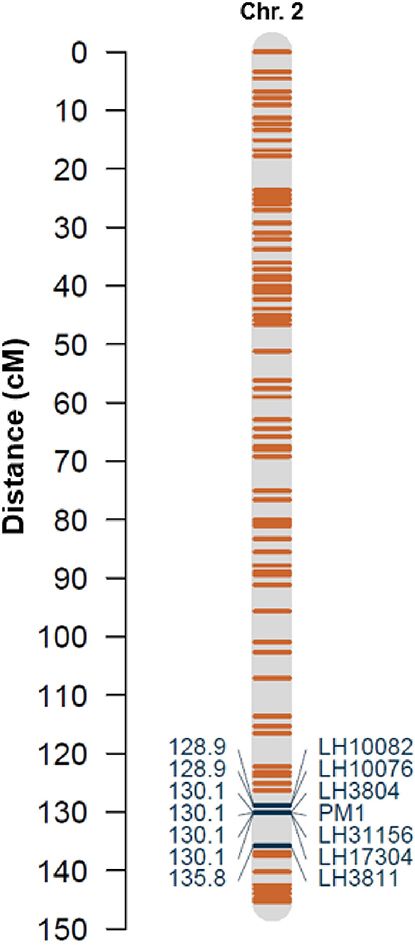
FIGURE 3. Linkage map describing locations of selected markers on chromosome 2 (based on “CBDRx” chromosome naming). Resistance gene PM1 co-localizes with SNP markers LH3804, LH31156, and LH17304 on chromosome 2.
Validation of PM_LH3804 as a Marker for PM1
The qPCR SNP genotyping assay PM_LH3804 classified F1 progeny from the “PNW39” × “Jumping Jack” population in two distinct clusters representing the susceptible (AA) and resistant (AG) individuals (Figure 4A). The SNP genotyping assay also confirmed that the resistant parent “PNW39” was heterozygous for the resistance allele. None of the individuals were classified as “GG,” in accordance with hypothesized inheritance patterns.
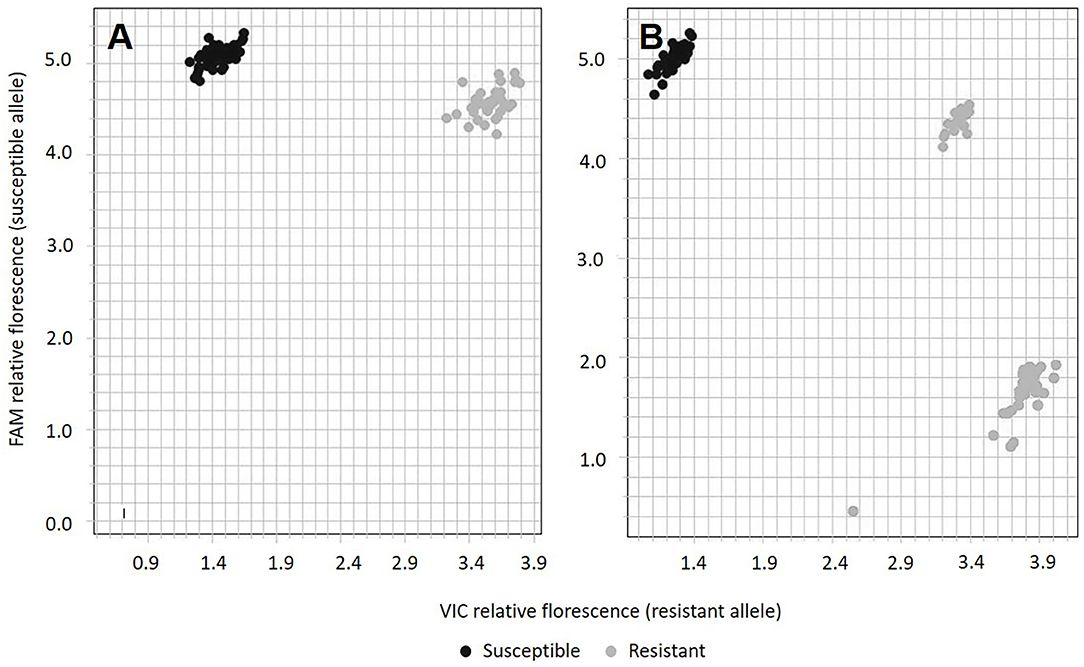
FIGURE 4. Allelic discrimination plot from SNP assay PM_LH3804 using (A) the parents and 89 F1 progeny from “PNW39” × “Jumping Jack” and (B) 96 F1 progeny from “PNW39” × “Dwy1337.” Black dots represent individuals with the susceptible phenotype and gray dots represent individuals with the resistant phenotype.
PM_LH3804 assay results from the “PNW39” × “Dwy1337” cross were unexpected insofar as a third genotype (representing “GG”) was indicated in the SNP analysis (Figure 4B) despite homozygous resistance being unlikely in this population. All susceptible progeny were scored as “AA,” but two genotype clusters represented the resistant progeny (19 individuals with “AG” and 30 individuals with “GG”) (Figure 2B). Despite the anomaly, addressed further in the discussion section, no recombinants were observable in this second population.
Local Reference Genome Annotations Place LH3804 Within a Cluster of R Genes
Primers Mildewey_1F and Mildewey_1R were used to amplify and sequence a 641 bp fragment surrounding the LH3804 locus using DNA from the resistant parent “PNW39” (nucleotide sequence supplied as Supplementary Data). MegaBLAST and BLASTn searches of the NCBI database with the built consensus sequence confirmed the BLAST results of the probe alone as being located on chromosome 2 of CBDRx and further resulted in hits to several predicted, putative, and probable disease resistance proteins on the same chromosome. A wider search of the region was conducted of the annotated CBDRx genome and a cluster of 10 putative disease resistance proteins were identified in the area surrounding the LH3804 locus, of which all contained N-terminal coiled-coil (CC) and nucleotide binding arc (NB-ARC) domains; two genes also contained leucine-rich repeats characteristic of plant R genes (Table 2). Three additional annotations were observed for pentatricopeptide repeat-containing proteins (Table 2), which are not usually implicated directly in disease resistance, but are assumed to share similar evolutionary histories with R genes and are typically found in clusters in the vicinity of R genes (Hernandez Mora et al., 2010).
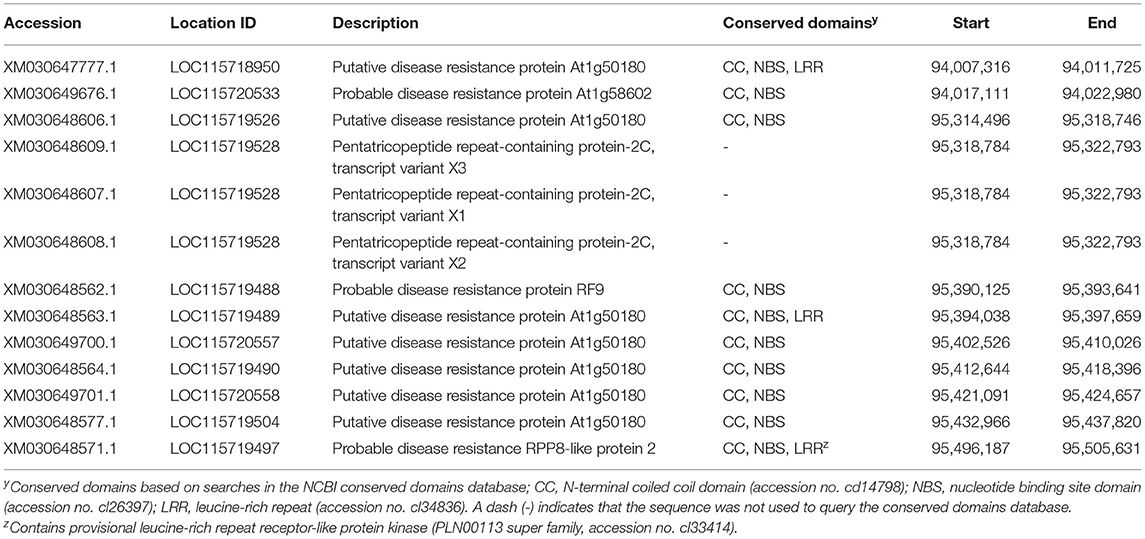
TABLE 2. Annotated resistance-like genes on chromosome 2 of the Cannabis sativa “CBDRx” genome surrounding the location of the LH3804 locus.
Discussion
Designation of PM1
Here we provide the first documentation of a powdery mildew resistance gene in C. sativa. Resistance observed among heterozygotes confirms the dominant behavior of the gene. Importantly, the integrity of the resistance is upheld in multiple genetic backgrounds and remains effective through plant maturity.
The formal name PM1 (Powdery Mildew-1) will be assigned to the R gene identified in this study. Among nearly 10,000 high-quality SNP markers surveyed throughout the genome, a single point mutation never broke linkage with 185 recombinant progeny. Using the annotated “CBDRx” genome, we have identified 10 possible candidate genes near the LH3804 locus which are not of the multigenic mlo type, but of the single dominant resistance R gene type. Specifically, we mapped PM1 to a region rich in genes which contain nucleotide-binding site (NBS) and leucine-rich repeat (LRR) domains. NBS-LRR R genes are known to code for pathogen effector detectors, are often found in tightly linked clusters, and in some cases, mediate programmed cell death as a defense response (Jones and Dangl, 2006; McHale et al., 2006). NBS or NBS-LRR type resistance proteins have been implicated in resistance to powdery mildew in species such as grape (Vitis vinifera) (Goyal et al., 2020) and wheat (Triticum aestivum) (He et al., 2018); nonetheless, there are few instances where the literature has definitively identified NBS-LRR genes as responsible for resistance to powdery mildew. NBS-type proteins have been identified as possible candidates for resistance to powdery mildew in hops (Kozjak et al., 2009; Padgitt-Cobb et al., 2019), but have not definitively been determined as is the case in the current study. At this point, we do not know which of the genes identified in the annotated “CBDRx” genome are responsible for resistance to the tested population of G. ambrosiae, or if there is an additional gene in “PNW39” not present in “CBDRx” that is linked to PM1 and conferring resistance. Determining the exact sequence among many candidate genes would require further study of the linkage block representing a ~2Mb chromosome segment.
Linkage mapping with 2,051 genome-wide chromosomal bins places PM1 on the distal end of the long arm of chromosome 2 based on the most current version of the “CBDRx” genome (accession no. LR213632.1). This positioning is of practical breeding importance; introgression with minimal linkage drag will be more feasible due to generally higher recombination frequency near telomeric ends. Investigation into the physical placement of the SNP loci used in linkage mapping, as determined by searches of the “CBDRx” genome, indicates that a large part of the chromosome on which PM1 is located displays low recombination density; of the 96 Mb long chromosome, more frequent recombination events are limited to ~15 Mb on each of the distal ends (Supplementary Table 3). Our results corroborate those reported by Kovalchuk et al. (2020) who show that chromosomes 1, 2, 3, and 4 of the C. sativa cultivar “Finola” exhibit unusually large proximal segments of low recombination. BLAST searches of the “Finola” genome have indicated that locus LH3804 and the remainder of the loci on our linkage map are located on chromosome 3 of “Finola” (accession no. CM011607.1) and is highly syntenic with chromosome 2 of “CBDRx.” There is no current consensus regarding chromosome numbering in C. sativa, therefore it is possible that chromosome 2 from “CBDRx” and 3 from “Finola” are likely describing the same chromosome.
Utility of PM_LH3804 in Molecular Breeding
Only one qPCR-based marker, derived from LH3804, was developed and confirmed in multiple genetic backgrounds. It is sometimes useful to incorporate flanking loci (e.g., LH10082, LH10076, LH3811) into marker-assisted selection methods to reduce the likelihood of error stemming from undetected recombination events; however, the 6.9 cM spanning the smallest interval (one marker bin on each side of LH3804) is inadequate for high-throughput screening and would also likely confer low utility during association mapping in diversity panels. Two additional markers, LH17304 and LH31156, co-segregated with the resistance locus but are accompanied by phasing problems as they are types D2.15 and B3.7, respectively. The best marker at this point is qPCR assay PM_LH3804, having maintained perfect linkage among 185 recombinant F1 progeny with accurate allele phasing in two distinct genetic backgrounds. Furthermore, since locus LH3804 was determined to be located within the coding region of a putative R gene, and since a search of the genomic region surrounding locus LH3804 did not identify any additional disease resistance gene clusters, this marker is likely to be situated in the gene cluster in which PM1 resides and is therefore unlikely to become unlinked to PM1.
PM_LH3804 assay performance in the “PNW39” × “Jumping Jack” population met postulated segregation, with the heterozygous cluster representing resistant progeny and the homozygous cluster representing susceptible progeny. Assay results in the second population, “PNW39” × “Dwy1337,” were more difficult to interpret. All susceptible progeny were scored in the same homozygous cluster, but the resistant progeny were split into two groups consisting of both the expected heterozygous cluster and an unexpected cluster for homozygous resistance. One plausible explanation is the presence of a third allele or a deletion event in the “Dwy1337” genome which would not be amplified in the qPCR assay designed for one of the two allele possibilities discovered by the SNP array. In this scenario, heterozygous individuals could erroneously be scored as homozygous for the resistance allele. While DNA from the original “Dwy1337” pollen donors are not available (and alone would not provide a sufficient answer), this hypothesis was explored further by screening several “Dwy1337” families derived from single-seed descent of original susceptible parental material. All individuals tested (n=32) were either homozygous susceptible or failed to amplify, further suggesting the presence of a third allele not being detected by the SNP genotyping assay. Sanger sequencing was performed on several of the progeny from this population using the Primers Mildewey_1F and Mildewey_1R, and several redesigns of these primers in close proximity, yet provided no clear indication of a third allele that would not be amplified by the PM_LH3804 assay. These results may support the deletion event theory, or perhaps indicate another sequence variant that cannot be amplified using the end-point primers that were tested. Despite this anomaly, PM_LH3804 accurately detected the resistance-associated allele in all tested progeny with no observable recombination events.
Considering the scenario of low linkage disequilibrium in global C. sativa germplasm (Onofri and Mandolino, 2017), PM_LH3804 is unlikely to be diagnostic of the resistance gene PM1 across all genetic backgrounds. Follow-up studies that aim to increase the resolution of this chromosomal region will undoubtedly lead to a more perfect marker. Furthermore, it is likely that additional studies will elucidate new genomic regions associated with powdery mildew resistance in diverse C. sativa accessions.
The State of Powdery Mildew R Gene Deployment in C. sativa
While disease pressure is anecdotally ubiquitous in both commercial and hobbyist growing conditions, comprehensive surveys of powdery mildew on C. sativa are not available in the public domain. R genes have almost certainly been deployed sporadically, but the literature also lacks documentation of their history, use, or any level of genetic characterization. Without allelism tests or genetic markers to compare the relatedness of R genes in cultivated varieties, it is challenging to determine how many R genes are currently in use or available in C. sativa germplasm. Quantitatively inherited resistance has been observed in recombinant inbred C. sativa populations (P.D. Mihalyov, unpublished data), but the complexity of marker development for polygenic resistance is likely to make the documentation of vertical R genes more prominent in scientific literature moving forward. Given that partial horizontal resistance can be more effective against diversified disease pressure, both strategies should be considered for the development of broadly durable cultivars (Pilet-Nayel et al., 2017).
Understanding the genetic variation and host specificities of experimental powdery mildew inoculum will be essential for the interpretation of future R gene studies in C. sativa. While G. ambrosiae was the causal agent of disease in this study, the R gene described herein may be ineffective against geographically or taxonomically diverse powdery mildew isolates. For example, C. sativa has also been found to be a natural host of the hop powdery mildew species Podosphaera macularis (Weldon et al., 2020; Bates et al., 2021) also report the cross-infectivity of P. macularis on hemp in controlled pathogenicity trials, however, did not report a naturally occurring infection) and it is unknown whether PM1 is effective against this species. Of equal importance will be the documentation of current and novel powdery mildew race emergence and their interactions with known R genes. After a series of genetically stabilized R gene differentials is compiled, coupled with screening their disease responses against a comprehensive panel of isolates, a robust strategy for the deployment of effective and enduring resistance may be implemented.
Data Availability Statement
All data are included as Supplementary Material.
Author Contributions
PDM and ARG analyzed the data, developed the tables and figures, and wrote the paper. PDM conceived the idea, developed the populations, and conducted phenotyping experiments. ARG designed and validated the qPCR assays, performed genomic alignments and searches, and conducted taxonomic pathogen classification. All authors contributed to the article and approved the submitted version.
Funding
Funding for this project was provided by Dewey Scientific and Oregon CBD.
Conflict of Interest
PDM is employed by Dewey Scientific. ARG is employed by Oregon CBD.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Jordan Zager and the staff of Dewey Scientific, and Seth Crawford, Eric Crawford, and the staff of Oregon CBD for their continuing support of exploratory breeding projects. Many thanks to Kelly Vining for her help with gene annotation searches and pre-submission review. We also thank Mark Lange, Kelly Vining, Jordan Zager, and Seth Crawford for their pre-submission reviews. Lastly, we are grateful to Philippe Henry, Anders Gonçalves da Silva, and Timothy Harvey at Lighthouse Genomics for their sequencing tools that made this research possible.
Explore Original Journal Here: https://www.frontiersin.org/articles/10.3389/fagro.2021.720215/full#B16